For many months, a team of biologists from the Salk Institute have been using SkyPilot to conduct research on the cloud on a daily basis.
In this guest user post, Dr. Hanqing Liu from the Ecker Lab shares their experiences. Cross-posted from the original post on Medium.
Modern genomic technologies generate vast amounts of data that necessitate significant computational resources for processing. Although cloud platforms are ideally suited to manage such immense computational demands, they remain underutilized in the biology field. This disparity primarily stems from the limited understanding of cloud technology and the daunting initial learning curve associated with setting up a cloud environment. Fortunately, several months ago, we at Salk Institute’s Ecker Lab added the open-source SkyPilot library from UC Berkeley to our toolkit. With SkyPilot’s assistance, we swiftly adapted our workflow to analyze extensive single-cell whole mouse brain atlas datasets entirely on the cloud.
In this post, we will describe why and how we completed this large genomic project using the cloud and SkyPilot. By the end of the post, other biologists and bioinformaticians should be able to understand the benefits of our cloud-based approach, as well as use the concrete examples we give to easily start using the cloud for their own research.
Background: Building a detailed parts list for an entire mammalian brain.
The brain is one of the most complex structures in the known universe. With each human brain comprising billions of cells wiring through trillions of synapses, the question arises: how can neuroscientists efficiently unravel the immense complexity of brain cells? A potential answer lies in the revolutionary single-cell technologies, which have empowered the development of cell atlases for entire organs, akin to a detailed parts list for a sophisticated machine.
In our recent bioRxiv preprint (Fig. 1), we presented a cutting-edge single-cell genomic dataset encompassing the entire mouse brain, a widely used animal model for investigating the fundamental principles of mammalian neural systems. This study comprises multiple datasets containing over 10,000,000 in-depth single-cell profiles (generated by us and other collaborators) associated with various molecular modalities crucial to brain function, such as RNA, DNA modification, and the 3D structure of DNA. Each included dataset involves the generation of hundreds of TBs of sequencing data.
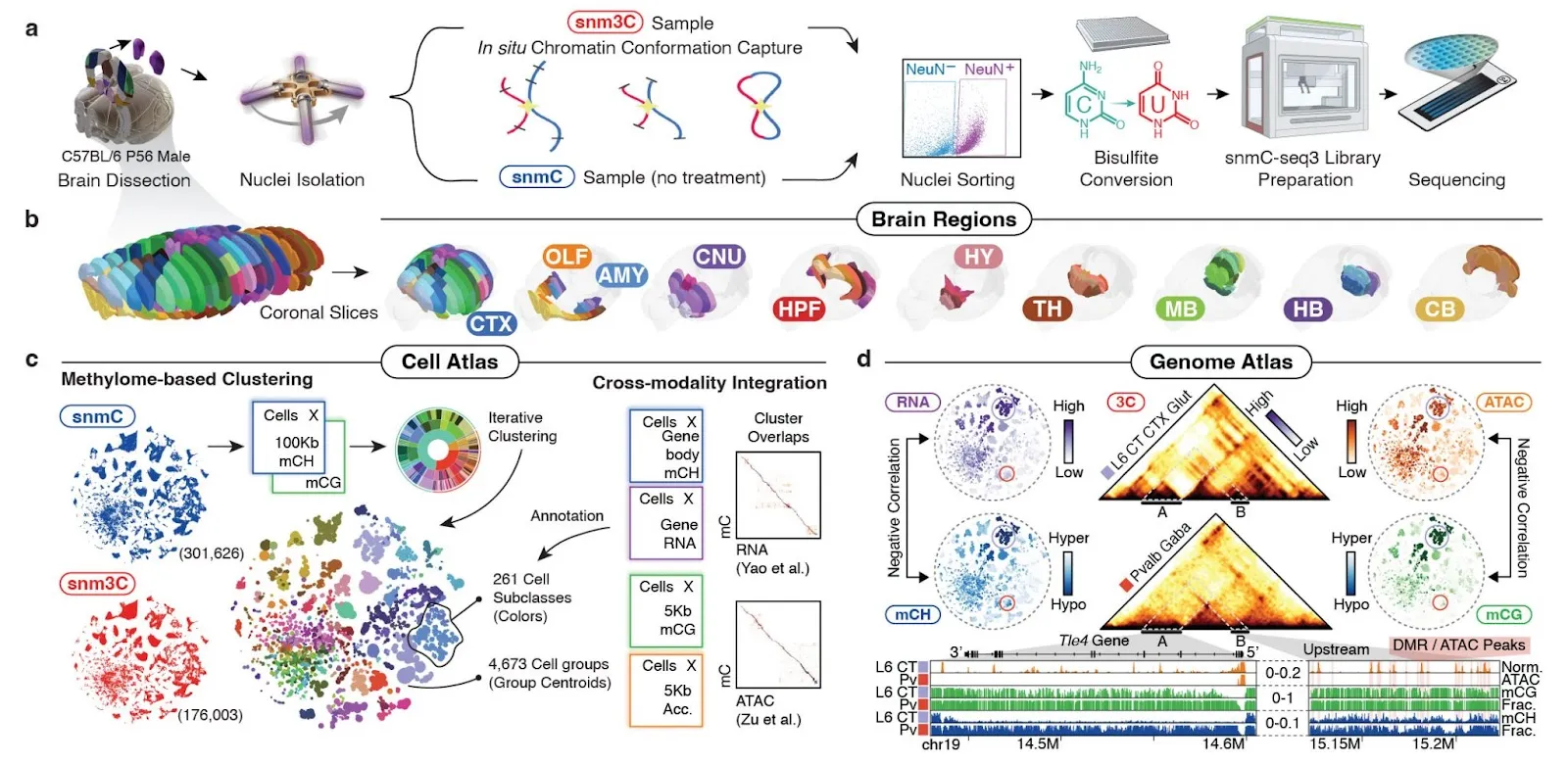
General Cloud Workflow with SkyPilot
Although comprehensive biological analyses are beyond the scope of this blog post and are detailed in our preprint, we will concentrate on outlining a daily workflow for all our investigations. Our general workflow comprises four parts, each mapped to an out-of-the-box concept in SkyPilot:
- Resources: Find an appropriate computational resource on a certain region and cloud;
- Data (file_mounts): Read/write tons of data, existing code, and genome reference files from/to cloud object storage buckets;
- Setup: Install dozens of bioinformatic packages;
- Run: Start a Jupyter server for interactive explorations or run a Snakemake (a popular bioinformatic pipeline package) command for batch pipelines.
We visualize this general process to demonstrate how SkyPilot significantly simplifies and enhances our cloud-based productivity, using a single YAML file containing 30 lines of code (Fig. 2).

Two use cases from actual analysis
Additionally, we present two specific examples below for readers interested in the actual biological analyses being conducted. If you have not used SkyPilot before, check out the SkyPilot “installation” and “quick start” in the documentation first.
Case I: Interactive exploration with Jupyter Notebooks
Similar to many data science projects, the initial phase of our analyses is often exploratory, necessitating access to the complete dataset using Jupyter Notebooks. In particular, a common task in single-cell data analysis involves conducting Leiden clustering on a cell-by-feature matrix. This clustering analysis comprises several preprocessing and preparation stages. Upon completing the clustering process, it is crucial to visualize the outcomes and conduct subsequent differential analyses to comprehend and characterize the cell clusters.
These tasks have several requirements on the computational environments, which can be hard to manage manually:
- Resources: Our demands constantly change based on the dataset size, ranging from a small VM (e.g.,
n2d-standard-16) to the largest VM possible (e.g.,n2d-highmem-96). - Data: The analysis may involve hundreds of files across multiple buckets. Directly copying all of them to the VM’s persistent disk is highly cumbersome.
- Setup: With new tools continually emerging, we want to test various options and document the installation steps for reuse on different VMs.
These requirements can be effortlessly addressed using a SkyPilot YAML file (Fig. 2). Let’s start with a basic example and modify it incrementally:
Create a VM with a very basic YAML file
Here is a minimum YAML file to start a small (n2d-standard-16) VM which is given access to the analysis and data buckets and starts a Jupyter server:
# cluster_vm.yaml
resources:
cloud: gcp
instance_type: n2d-standard-16
disk_size: 128
file_mounts:
/data:
source: gs://project-data-bucket/
mode: MOUNT
/home/analysis:
source: gs://my-analysis-bucket/
mode: MOUNT
setup: |
pip install --upgrade pip
conda env update -f /home/analysis/env.yaml
run: |
nohup jupyter-lab --no-browser --port 8888 &
To start a VM named “clustering” with this command, simply run in the shell:
$ sky launch -c clustering cluster_vm.yaml
In just about a minute, we have a VM up and running, ready for clustering analysis! This magic line brings several remarkable benefits:
- The VM has access to two of my buckets, one (
gs://project-data-bucket/) with large data files, the other (gs://my-analysis-bucket) containing my daily analysis folders. These buckets are mounted to the VM’s file system, enabling access to them like any other local files on the system. - All packages listed in
/home/analysis/env.yamlare automatically installed during the VM setup. It’s worth noting that/home/analysis/env.yamlis actually located in the remote bucketgs://my-analysis-bucket/env.yaml. Thanks to the file mounts in (1), we don’t need to download it before using it! - A Jupyter Notebook server is running once the VM starts. Moreover, SkyPilot also set ups an SSH connection between the local machine and the new VM, so we can directly SSH into the VM or create an SSH tunnel to the Jupyter Notebook server simply by using the convenient VM name that we specified (rather than copy-pasting IPs).
$ ssh clustering # enter the VM
$ ssh -L 8888:localhost:8888 clustering # or create an SSH tunnel to the running Jupyter server
After finishing our analysis, turning off the VM is also simple:
$ sky stop clustering
If we are afraid of forgetting to turn off the VM (often times I will), an even better solution is to use the autostop feature:
# this will turn off the VM after 4 hours.
$ sky autostop clustering -i 240
Next time (typically next morning) when we want to continue the analysis, just repeat the sky launch command above (which will run the setup) or sky start (which will not rerun the setup) and everything will come back!
What if I need more resources?
Simply ask for different VM types in the resources section in the YAML file. For example, if we want a much larger machine, several monster GPUs ($$$), and more disk space, just ask for it:
resources:
cloud: gcp
instance_type: n2d-highmem-64
disk_size: 512
accelerators: V100:4
We can even ask for resources on a different cloud provider (e.g., GCP, AWS, Azure, Lambda Cloud), as SkyPilot is designed to work on any cloud. We can also let SkyPilot help to choose the cheapest solution.
What if I have a new project’s data in another bucket?
Commonly, our daily analysis involves multiple projects. Moving files around can be painful and error-prone. With SkyPilot, we don’t need to do that. Just add another item in the file_mounts section in the YAML file:
file_mounts:
/new_data/:
source: gs://new-project-data-bucket/
mode: MOUNT
/reference/:
source: gs://genome-reference-bucket/some/reference/dir/
mode: COPY
# followed by other file mounts, as many as you need.
In addition to the MOUNT mode, we can also COPY the files to this VM (automatically!), which might be ideal for some frequently visited files (e.g., genome reference). Check out more options in the documentation.
What if I need to install new tools?
Adding new tools during the VM setup is easy. We can record all installation commands in the setup section of the YAML file:
setup: |
… # previous install code
pip install a_new_cool_package
# or any git clone, R installation commands
Remember that everything under setup is just shell commands. It will be executed automatically whenever we use sky launch to start the VM.
Case I summary
In this case, we employ the versatile SkyPilot framework to meet our daily requirements for conducting interactive data analysis using the Jupyter Notebook server. In practice, creating a reusable SkyPilot YAML file takes approximately 5 minutes, while starting a VM requires just about 1 minute. Thanks to this efficient framework, setting up a VM in the morning becomes faster than waiting for a cup of coffee from the espresso machine.
Case II: Batch scale-up with Snakemake pipelines on spot-VM.
Typically, the goal of exploratory data analysis is to establish a refined analysis pipeline that can be applied to orders of magnitudes larger datasets. In genomic data analysis, such pipelines are often composed within a Snakefile, which is subsequently managed by the Snakemake workflow management system for execution through a single command. An example of such a pipeline, which processes raw sequencing data and generates analysis-ready files, can be found here.
Since many of these pipelines are resource-intensive, cost becomes a primary factor deterring biologists from utilizing cloud services. For instance, our current mapping pipeline takes approximately 1 CPU hour to process a single cell (its slow nature is due to each cell containing several million reads). Given that our whole-mouse-brain dataset comprises around 0.5 million cells, merely mapping the data (one of the numerous analysis steps) could amount to tens of thousands of dollars in expenses.
An excellent solution for reducing the cost of batch jobs in the cloud is to use spot (or preemptible) machines. For example, on the Google Cloud Platform, spot VMs are typically several times cheaper than on-demand machines. However, the flip side is that spot VMs can be challenging to manage from an engineering standpoint. Due to their lower cost, cloud providers can shut down the VM anytime without notice in a process called preemption. While it is possible to restart the machine following preemption, the workload might need to restart, and intermediate files may become corrupted if not managed properly.
SkyPilot manages spot jobs automatically with ~6x cost saving
Fortunately, SkyPilot addresses this engineering challenge. With the same YAML file from before, we can start a “managed spot job” using a single sky spot launch command. SkyPilot will manage the spot VMs, automatically recover from preemptions, and even fail over to different regions or cloud platforms if spot resources are unavailable.
Here is an example YAML file we use to run a mapping job on a spot VM. It looks identical to the previous interactive jobs, except that in the run section in the YAML file, we will execute a snakemake command to perform some batch jobs (mapping single-cell reads to the genome).
# mapping_vm.yaml
resources:
cloud: gcp
instance_type: n2d-standard-64
disk_size: 256
file_mounts:
/input:
source: gs://sequencing-raw-data-bucket/
mode: MOUNT
/output:
source: gs://mapped-data-bucket/
mode: MOUNT
/ref:
source: gs://genome-reference-bucket/
mode: COPY
setup: |
pip install --upgrade pip
conda env update -f /ref/mapping_env.yaml
run: |
snakemake --snakefile SNAKEFILE --directory DIR --cores 32
We can execute the mapping with the single sky spot launch command in shell:
$ sky spot launch -n mapping mapping_vm.yaml
The crucial distinction is that an on-demand n2d-standard-64 VM costs $2.70 per hour, while the spot n2d-standard-64 VM costs $0.41, namely 6.5x cheaper (at the time of writing, in the us-west1 region). Although the VM is subject to preemption, SkyPilot will automatically restart it after preemption and resume the workload (in our case, the Snakemake command) (Fig. 3). Once Snakemake resumes, it can restart the pipeline from its own checkpoints, further ensuring the workflow continuity (thanks to Snakemake’s checkpoint capabilities as well).
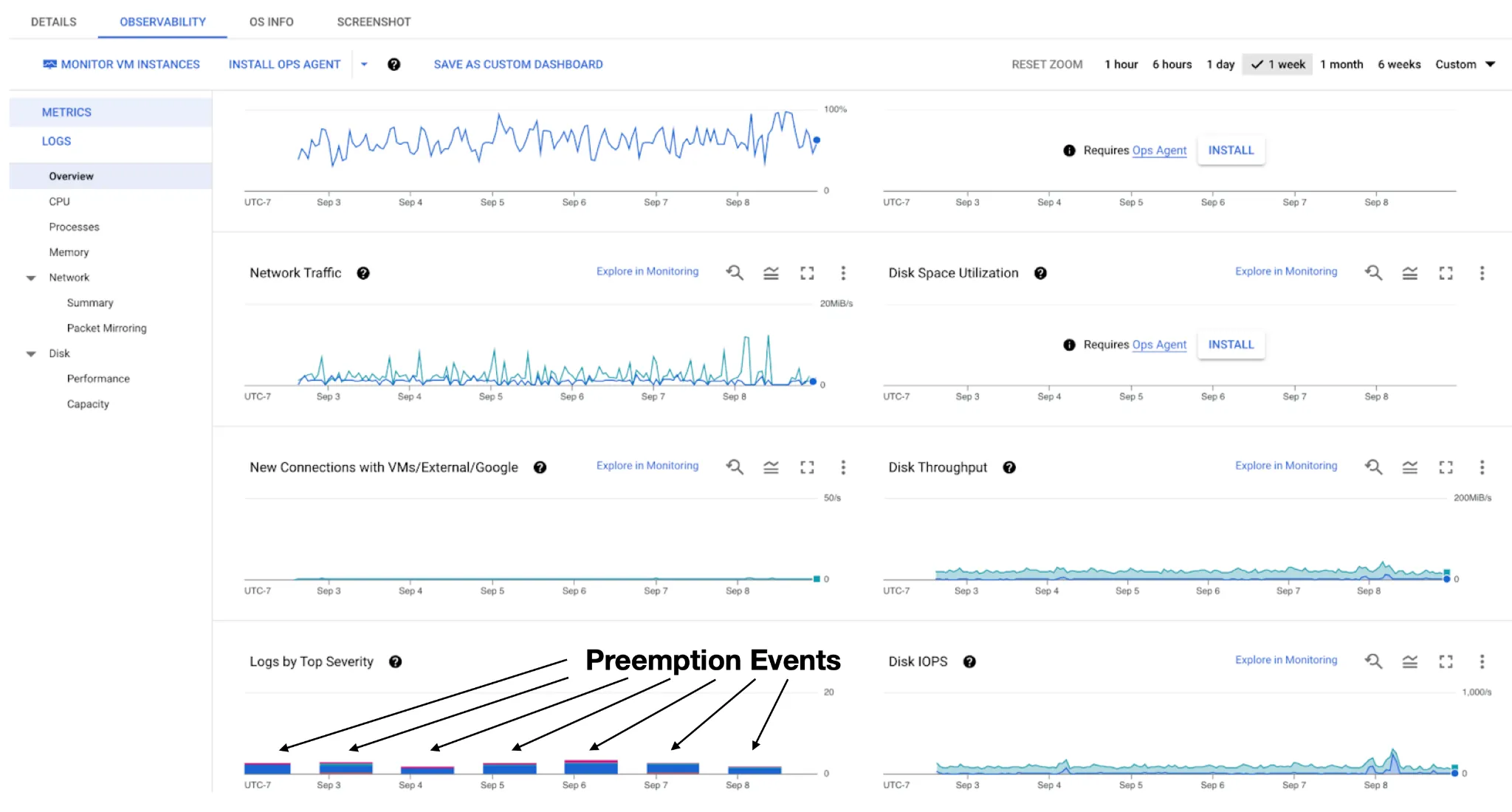
Case II summary and our five-months experience
In this case, we introduce the Spot feature of the SkyPilot framework, which is essential to make the Spot-VM usable for daily, resource-intensive batch jobs. Over the past five months, we have extensively utilized SkyPilot to manage Spot-VMs (Fig. 4), resulting in an estimated 5.7X cost savings compared to using on-demand VMs.
Besides, if the same computational work were to be executed on our on-prem HPC facilities (in-house server + Stampede2 Supercomputer at the Texas Advanced Computing Center), it would’ve taken approximately 15 months (wall time) to complete. The extended duration of on-prem HPC is attributed to various factors, which we will discuss in the subsequent section. Achieving a 3X wall time speed up is vital for the productivity and timely completion of large-scale projects, such as constructing a comprehensive mouse brain atlas.

Why did we move to the cloud?
Prior to adopting cloud computing, we have utilized on-premises computational resources, as well as low-cost or complimentary high-performance computing (HPC) systems at supercomputer centers. However, after years of managing ever-expanding genomic datasets on-premises, we have encountered numerous constraints that are more effectively resolved through cloud-based solutions. In Table 1, we outline key factors that have influenced our increasing reliance on cloud computing, both now and in the foreseeable future.
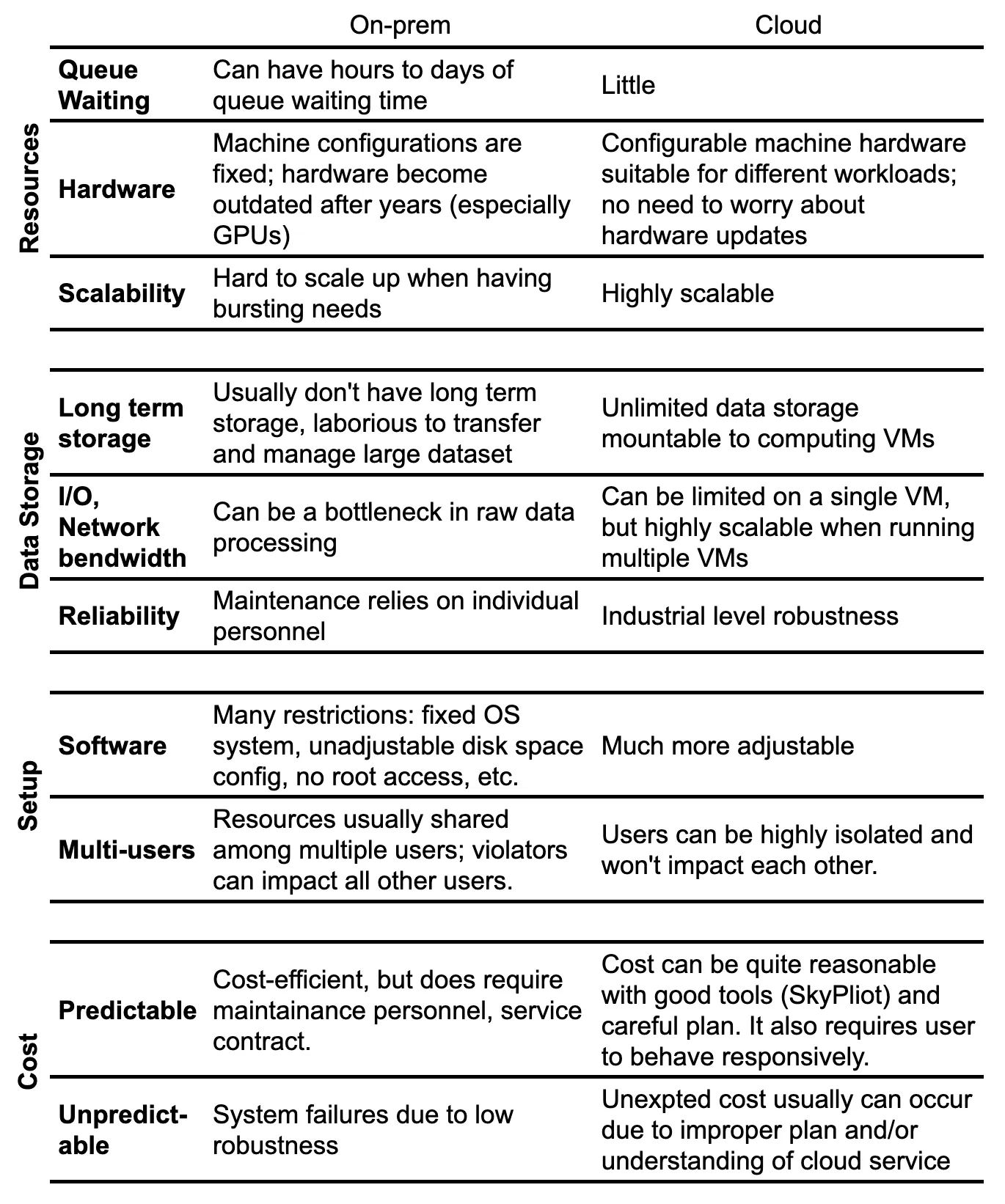
What does SkyPilot bring to bioinformaticians?
SkyPilot is a versatile framework for running any jobs on any cloud. As bioinformaticians, we particularly appreciate this framework for two primary reasons:
- Cost-effectiveness: SkyPilot is engineered to optimize cloud computing expenses.
- Time-efficiency: The framework enables seamless access to extensive cloud resources, requiring minimal learning time.
The field of bioinformatics is poised for a rapid expansion in data volume and computational requirements in the years to come. For instance, upon the completion of our mouse brain atlas projects, the next phase of the NIH BRAIN Initiative aims to support the development of a human brain atlas. Even if you are not generating massive datasets yourself, it is highly likely that you will require substantial computational power to effectively address your research questions using these extensive reference atlases.
As such, do not hesitate to harness the power of SkyPilot and cloud platforms for your most demanding computational tasks today!